- Home
- Genomes
- Genome Browser
- Tools
- Mirrors
- Downloads
- My Data
- Projects
- Help
- About Us
EBOLA AND MARBURG VIRUS CONSERVATION
Today we will be using BLAT to compare the Marburg virus genome to UCSC's display of the ebolavirus genome assembly. The Marburg virus is related to the Ebola virus, both in the family Filoviridae. Thus, in this example, I will illustrate how we can use BLAT to compare conservation between the two viruses and discuss why certain parts of the two viruses are conserved and ways to observe this.
Let's start by first opening the UCSC genome browser main page by typing genome.ucsc.edu into our search bar and clicking on the link titled "Genome Browser." From here, we can see a list of all the different organism assemblies currently available. If we scroll all the way down to the bottom under "viruses," we can locate the ebolavirus assembly. Clicking on this will refresh the page to the ebolavirus assembly. Clicking on "GO" from this screen will bring us to the ebolavirus's main session window. This will be our home screen from now on.
Here on
 our main session window,
we can first observe a few things about the ebolavirus:
our main session window,
we can first observe a few things about the ebolavirus:
- The genes expressed by the ebolavirus and the purpose each gene serves (by clicking on the gene link)
- 160 virus sample strains and the conservation among them, including the relatedness of viruses to others in the same outbreak (Figure 1)
- Various other data sets that can be viewed in different display modes.

Figure 1. Browser view of the Ebola virus genome. The clustering of viruses from different outbreaks is evident in the coloring.
While we can immediately dive into the conservation analysis, let's first begin with the BLAT search/comparison between the Marburg and ebola viruses. If we look at the top blue bar under Tools, we will find the BLAT tool as our first choice. Clicking here we will be brought to the BLAT tool main page, where we can tweak the options and enter our search DNA, RNA, or protein sample into the text box. For this analysis, I will be using a Marburg virus genome from GenBank which can be found at NCBI. After copying and pasting this genome sequence into the box, we can tweak our search preferences or just simply click on "Submit."
After clicking "submit," we are brought to the BLAT search result page. At first glance, we can notice most matches of roughly 20 or fewer bases but the best match is at the top of the list: 1300+ bases. If we click on the associated "browser" link, we are brought to the exact site of this match within our main session.
Now that we are back to our main session, let's click on the top browser link, zoom out by 3x, and make a few observations (Figure 2). In the resulting session window, we can see beneath the blue NCBI Genes track, the BLAT match of the Marburg virus we searched with.

Figure 2.
http://genome.ucsc.edu/s/education/ebolav_blatsearch.
Browser view of Marburg virus blat footprint on the Ebola virus genome. Purple arrow:
left end of BLAT alignment match.
In this session, we can see that the matches are two colors, purple (located at both ends of the long matching sequence, such as where the arrow is pointing and detached from the sequence as small isolated segments) and red. Clicking on the short BLAT matches we are brought to a color key page (Figure 3).

Figure 3. BLAT description page explaining colors.
Here we learn that purple is used where the sequence extends beyond the end of the alignment and red where the sequence and reference genome have different bases at this position.
Clicking on the back arrow in our browser window, we are brought back to the main session. Knowing what each color denotes, we can see that a large portion of our Marburg virus matches the ebolavirus genome, but varies in bases.
If we zoom in on a particular section of the red, we can directly observe this variation (Figure 4).

Figure 4a.
http://genome.ucsc.edu/s/education/ebolav_codon.
Zoomed-in section of the ebolavirus. Highlighted section to show main ebola sequence nucleotides.

Figure 4b. Bottom part of the previous figure with the same yellow highlight. Here a change of a single nucleotide affects the identity of the amino acid in strains distantly related to the reference.
The bases shown against the gray background of our BLAT Marburg virus probe ("Your Seq" near the top of the window), represent bases that do not match the ebola virus reference genome. Changes in the first and second nucleotides of a codon usually result in entirely new amino acids.
On a similar note, if we scroll down to the bottom of our 160 Strains Comparative Genomics data track, we can directly see that some outbreaks of Ebola have different amino acids at this location (Figure 4b).

Figure 5. Hovering your mouse over the "Protein Annotations" track, we can see the gene annotated RNA-polymerase gene L
Looking at our BLAT result, we can see that the main similarity between the Marburg virus and ebolavirus lies within the L gene (Figure 5) — an important part of the virus s functionality as it encodes the virus's transcriptase, integral to the virus's ability to replicate and survive. This supports the idea that these areas would be the region most likely to match because ebolavirus and Marburg virus are in the same family and replicate/spread in similar ways.
Now let's move on to our second observation, conservation within the ebolavirus genome. As we just saw, variation or conservation of amino acids among the 160 different strains is higher in certain areas than others. Again, this can be seen by scrolling down to the bottom of our 160 Strains Comparative Genomics data track.
Another data track we can use to observe conservation is the PhyloP data track which can be found near the top as a small varying thin pink line. The pink color in the Browser indicates that the data extends beyond the maximum value displayed in the track. In this view, the data are not very informative. We will improve on that below. Before progressing any further though, let's click on the zoom out 100x to be brought to nearly the full ebolavirus genome assembly. To see this example of gene conservation, let's highlight and zoom in on the gene "VP24" (Figure 6).
Figure 6. Highlight/Zoom-in window pop-up after highlighting the VP24 gene.
Doing so will bring us to this session window (Figure 7) with VP24 at nearly full screen-width:

Figure 7.
http://genome.ucsc.edu/s/education/ebolav_vp24.
VP24 gene zoomed in on the coding region. PhyloP data track shows conservation in
the protein-coding region. Data that extend beyond the display range are shown in
pink.
In Figure 7, we can observe the coding region of the VP24 gene, as well as the amino acids that encode this protein, shown at this resolution as alternating thin color changes. Within the upper middle of this session window, we can see the PhyloP score. PhyloP tracks and compares the conservation of 158 different virus strains in addition to two Marburg virus strains. Marburg is included in this dataset because it is related to Ebola.
However, to directly observe gene conservation, we will need to zoom in to directly see the varying PhyloP scores. To do this, let's right-click on the left tab next to the PhyloP track and click on "Configure PhyloP" (Figure 8):

Figure 8. Clicking on the gray left panel next to the PhyloP data track brings this drop-down menu.
Doing so will open the configuration window shown in Figure 9:
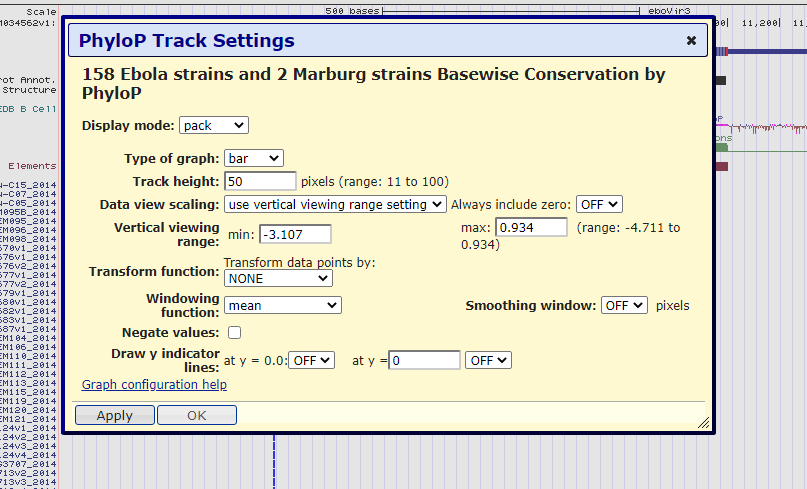
Figure 9. Pop-up menu to customize the PhyloP data track. This menu can adjust the size of the data track to make it more apparent and can be modified to include or exclude parts of the data.
Let's make a few adjustments to modify the track to make the data more apparent. First, let's change the display mode to full &mdash allowing for a larger, more readable graphic display. In addition, let's change the track height from 50 to 100. Next, let's change the vertical viewing range min from -3.107 to -5 and max from 0.934 to 5. In Figure 10, we can observe these changes. Finally, let's click on "Apply" and observe the changes we just made:
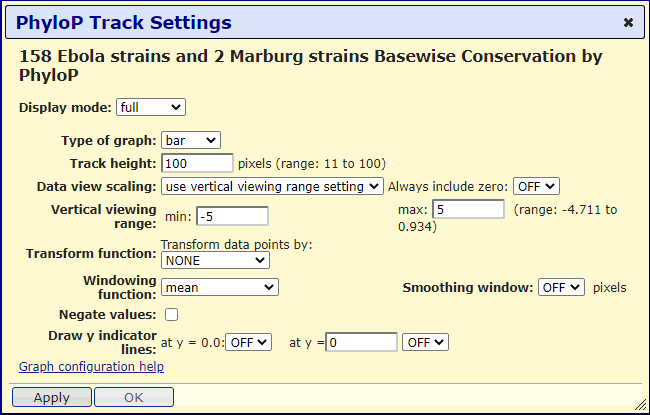
Figure 10. All PhyloP data track changes to make the data track easier to read.
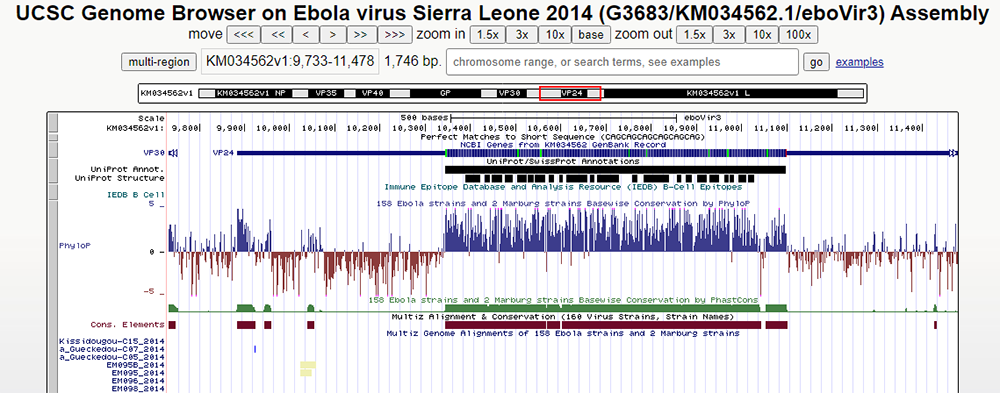
Figure 11.
http://genome.ucsc.edu/s/education/ebolav_phylop.
User-friendly display of the PhyloP data track after configuration modifications.
Notice in Figure 11 how our PhyloP data track is more obvious and is now presented in a lot more detail and user-friendly format. Now let's observe our genetic conservation at last!
To understand the PhyloP data track, we must understand the color-coding scheme as well as the numbers seen when hovering over the chart. The blue color signifies positive conservation, reporting the level that each nucleotide present within each amino acid is conserved/kept the same throughout all of the strains. Brownish red on the other hand signifies variation/variety, signifying a possible mutation or overall variation of nucleotides among the 160 strains.
We can zoom into a specific blue region and observe more directly genetic conservation at play (Figure 12):
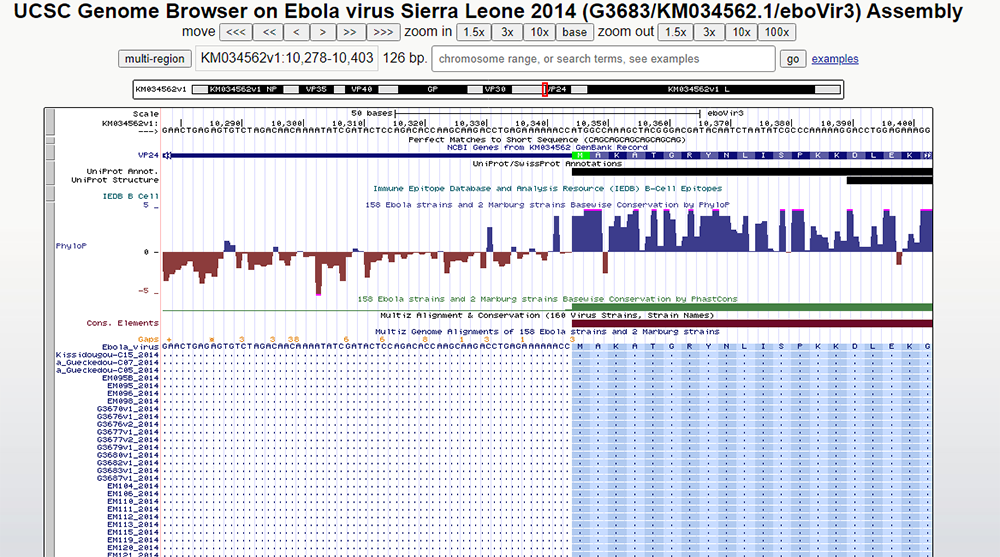
Figure 12.
http://genome.ucsc.edu/s/education/ebolav_pconserv.
Close zoom-in on the start codon area for the VP24 gene. Here we can directly observe the difference in conservation between nucleotides upstream (left) and downstream (right) from the methionine (green codon).
In Figure 12, we can see that the methionine amino acid is highly conserved as it's the start of the VP24 gene. Before this methionine, the conservation of nucleotides varies dramatically because these nucleotides are not encoding protein nor are essential for the coding of VP24. The small orange numbers indicate insertions in one or more of the strains relative to the reference.
The main point of this observation and the previous is the argument that conservation implies functionality. In areas not encoding amino acids (here, a 5'-untranslated region, UTR), we find that conservation is low. Furthermore, even though the Marburg virus we used in the BLAT query above did not align with the VP24 region, the PhyloP Conservation score indicates that VP24 is also a necessary component of Ebola. Marburg and Ebola have diverged in the VP24 region.
Written by Mateo Etcheveste, UCSC. Major: BS, Biomolecular Engineering