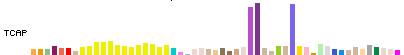
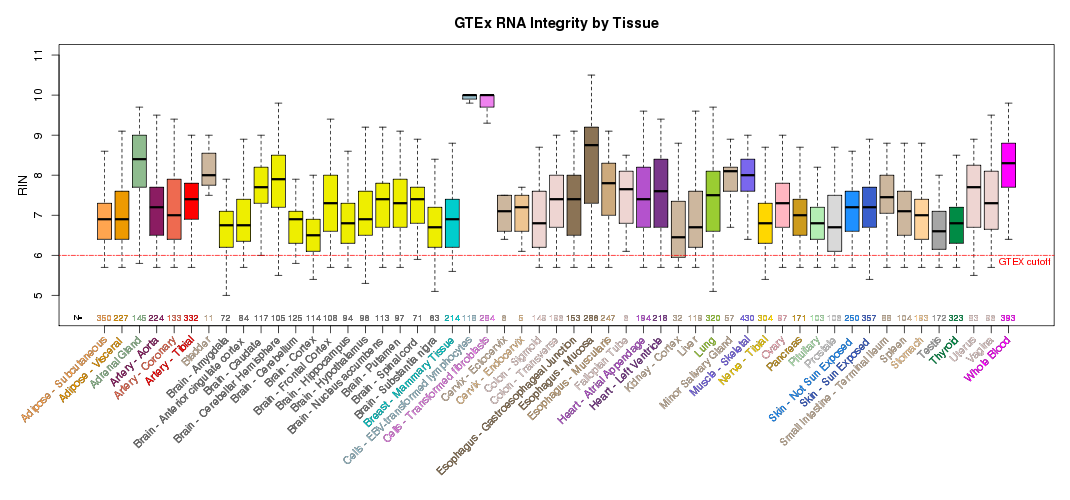
Description
The
NIH Genotype-Tissue Expression (GTEx)
project was created to establish a sample and data resource for studies on the relationship
between genetic variation and gene expression in multiple human tissues.
This track displays median transcript expression levels in 53 tissues, based on
RNA-seq data from the GTEx midpoint milestone data release (V6, October 2015).
To view the GTEx tissues in anatomical context, see the
GTEx Body Map.
Data for this track were computed at UCSC from GTEx RNA-seq sequence data using the
Toil
pipeline running the kallisto transcript-level quantification tool.
Display Conventions
In Full and Pack display modes, expression for each transcript is represented by a colored
bar chart, where the height of each bar represents the median expression level across all
samples for a tissue, and the bar color indicates the tissue.
The bar chart display has the same width and tissue order for all transcripts.
Mouse hover over a bar will show the tissue and median expression level.
The Squish display mode draws a rectangle for each gene, colored to indicate the tissue
with highest expression level if it contributes more than 10% to the overall expression
(and colored black if no tissue predominates).
In Dense mode, the darkness of the grayscale rectangle displayed for the transcript reflects
the total median expression level across all tissues.
Click-through on a graph displays a boxplot of expression level quartiles with outliers,
per tissue.
Methods
Tissue samples were obtained using the GTEx standard operating procedures for informed consent
and tissue collection, in conjunction with the
National Cancer Institute Biorepositories and Biospecimen.
All tissue specimens were reviewed by pathologists to characterize and
verify organ source.
Images from stained tissue samples can be viewed via the
NCI histopathology viewer.
The Qiagen PAXgene non-formalin tissue preservation product was used to stabilize
tissue specimens without cross-linking biomolecules.
RNA-seq was performed by the GTEx Laboratory, Data Analysis and Coordinating Center
(LDACC) at the Broad Institute.
The Illumina TruSeq protocol was used to create an unstranded polyA+ library sequenced
on the Illumina HiSeq 2000 platform to produce 76-bp paired end reads at a depth
averaging 50M aligned reads per sample.
Sequence reads for this track were quantified to the hg38/GRCh38 human genome using kallisto
assisted by the GENCODE v23 transcriptome definition. Read quantification was performed at UCSC
by the Computational Genomics lab, using the Toil pipeline. The resulting kallisto files were
combined to generate a transcript per million (TPM) expression matrix using the UCSC tool,
kallistoToMatrix. Average TPM expression values for each tissue were calculated and
used to generate a bed6+5 file that is the base of the track. This was done using the UCSC
tool, expMatrixToBarchartBed. The bed track was then converted to a bigBed file using the
UCSC tool, bedToBigBed.
The data in the hg19/GRCh37 version of this track was generated by converting the
coordinates from the hg38/GRCh38 track data.
Of the 189,615 BED entries from the original hg38 track, 176,220 were mapped over by transcript
name to hg19 using wgEncodeGencodeCompV24lift37 (~93% coverage).
Subject and Sample Characteristics
The scientific goal of the GTEx project required that the donors and their biospecimen
present with no evidence of disease. The tissue types collected were chosen based on their
clinical significance, logistical feasibility and their relevance to the scientific goal
of the project and the research community. Postmortem samples were collected from
non-diseased donors with ages ranging from 20 to 79. 34.4% of donors were female and
65.6% male.
Additional summary plots of GTEx sample characteristics are available at the
GTEx Portal Tissue Summary page.
Credits
Samples were collected by the GTEx Consortium.
RNA-seq was performed by the GTEx Laboratory, Data Analysis and Coordinating Center
(LDACC) at the Broad Institute.
John Vivian, Melissa Cline, and Benedict Paten of the UCSC Computational Genomics lab were
responsible for the sequence read quantification used to produce this track. Kate Rosenbloom
and Chris Eisenhart of the UCSC Genome Browser group were responsible for data file
post-processing and track configuration.
References
J. Vivian et al.,
Rapid and efficient analysis of 20,000 RNA-seq samples with Toil
bioRxiv bioRxiv, vol. 2, p. 62497, 2016.
GTEx Consortium.
The Genotype-Tissue Expression (GTEx) project.
Nat Genet. 2013 Jun;45(6):580-5.
PMID: 23715323;
PMC: PMC4010069
Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET et al.
A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project.
Biopreserv Biobank. 2015 Oct;13(5):311-9.
PMID: 26484571;
PMC: PMC4675181
Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM,
Pervouchine DD, Sullivan TJ et al.
Human genomics. The human transcriptome across tissues and individuals.
Science. 2015 May 8;348(6235):660-5.
PMID: 25954002; PMC: PMC4547472
DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G.
RNA-SeQC: RNA-seq metrics for quality control and process optimization.
Bioinformatics. 2012 Jun 1;28(11):1530-2.
PMID: 22539670; PMC: PMC3356847
|