ID:FIBA_HUMAN DESCRIPTION: RecName: Full=Fibrinogen alpha chain; Contains: RecName: Full=Fibrinopeptide A; Contains: RecName: Full=Fibrinogen alpha chain; Flags: Precursor; FUNCTION: Fibrinogen has a double function: yielding monomers that polymerize into fibrin and acting as a cofactor in platelet aggregation. SUBUNIT: Heterohexamer; disulfide linked. Contains 2 sets of 3 non-identical chains (alpha, beta and gamma). The 2 heterotrimers are in head to head conformation with the N-termini in a small central domain. SUBCELLULAR LOCATION: Secreted. TISSUE SPECIFICITY: Plasma. DOMAIN: A long coiled coil structure formed by 3 polypeptide chains connects the central nodule to the C-terminal domains (distal nodules). The long C-terminal ends of the alpha chains fold back, contributing a fourth strand to the coiled coil structure. PTM: The alpha chain is not glycosylated. PTM: Forms F13A-mediated cross-links between a glutamine and the epsilon-amino group of a lysine residue, forming fibronectin- fibrinogen heteropolymers. PTM: About one-third of the alpha chains in the molecules in blood were found to be phosphorylated. PTM: Conversion of fibrinogen to fibrin is triggered by thrombin, which cleaves fibrinopeptides A and B from alpha and beta chains, and thus exposes the N-terminal polymerization sites responsible for the formation of the soft clot. The soft clot is converted into the hard clot by factor XIIIA which catalyzes the epsilon- (gamma-glutamyl)lysine cross-linking between gamma chains (stronger) and between alpha chains (weaker) of different monomers. PTM: Phosphorylation sites are present in the extracellular medium. DISEASE: Defects in FGA are a cause of congenital afibrinogenemia (CAFBN) [MIM:202400]. This is a rare autosomal recessive disorder characterized by bleeding that varies from mild to severe and by complete absence or extremely low levels of plasma and platelet fibrinogen. Note=The majority of cases of afibrinogenemia are due to truncating mutations. Variations in position Arg-35 (the site of cleavage of fibrinopeptide a by thrombin) leads to alpha- dysfibrinogenemias. DISEASE: Defects in FGA are a cause of amyloidosis type 8 (AMYL8) [MIM:105200]; also known as systemic non-neuropathic amyloidosis or Ostertag-type amyloidosis. AMYL8 is a hereditary generalized amyloidosis due to deposition of apolipoprotein A1, fibrinogen and lysozyme amyloids. Viscera are particularly affected. There is no involvement of the nervous system. Clinical features include renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash. SIMILARITY: Contains 1 fibrinogen C-terminal domain. WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/FGA"; WEB RESOURCE: Name=SHMPD; Note=The Singapore human mutation and polymorphism database; URL="http://shmpd.bii.a-star.edu.sg/gene.php?genestart=A&genename=FGA"; WEB RESOURCE: Name=Wikipedia; Note=Fibrinogen entry; URL="http://en.wikipedia.org/wiki/Fibrinogen"; WEB RESOURCE: Name=SeattleSNPs; URL="http://pga.gs.washington.edu/data/fga/";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
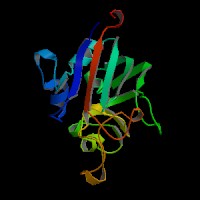
ModBase Predicted Comparative 3D Structure on P02671
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles